“The Sickle Cell Mutation or Malaria Resistance”
The Good: The Sickle Cell protein mutation leads to an abnormally shaped red blood cell to emerge not round but in a deformed sickle shape. The Sickle Cell mutation affects the beta-globin (HBB) gene and has also been found in at least two other pathways, including encoding hemoglobin S and C in the beta-globin. This mutation directly allows those affected to have a 29% – 93% survival rate of Malaria-1 because it did not recognize the mutated red blood cell proteins as food. Therefore, counter-intuitively, the misfolded sickle-shaped red blood cells provide a survival benefit that permitted millions of human beings from dying from the horrible disease of Malaria.
The reality: Sickle Cell is an inherited blood disorder or disease brought about by a genetic mutation that allows carriers to survive Malaria. The mutation is known to cause severe compilations in those that carry the trait. The disease can be anywhere from painful to deadly. Other side effects include Acute Chest Syndrome, stroke, learning disabilities, heart problems, kidney problems, and other degradative conditions, including anemia, pain, swelling of hands and feet, increased rates of infection, delayed puberty or growth, pulmonary hypertension, multiple organ damage, loss of body water in urine, blindness, leg ulcers, gallstones, priapism (obstruction of blood vessels in the penis), and other vision problems. Symptoms include fever, pale skin, yellowing (jaundice) eyes, stroke, trouble walking, confusion, and more. Sickle Cell also increases blood pressure and blood clots, especially during pregnancy increasing miscarriage, premature births, and low birth weights.-3 The mutation persists today as an endemic disease which means it cannot be removed from the genome and will persist in humankind, never to be “corrected” (removed).-2 Also, the mutation is associated with splenetic sequestration (pooling), and other disabilities and conditions.-3
As with all inherited genetic diseases, you’d expect natural selection to week out a gene that has such unpleasant consequences but with sickle cell disease, that doesn’t seem to be the case…Indeed, as of 2015, about 4.4 million people have sickle cell disease, while an additional 43 million have sickle cell trait.-3
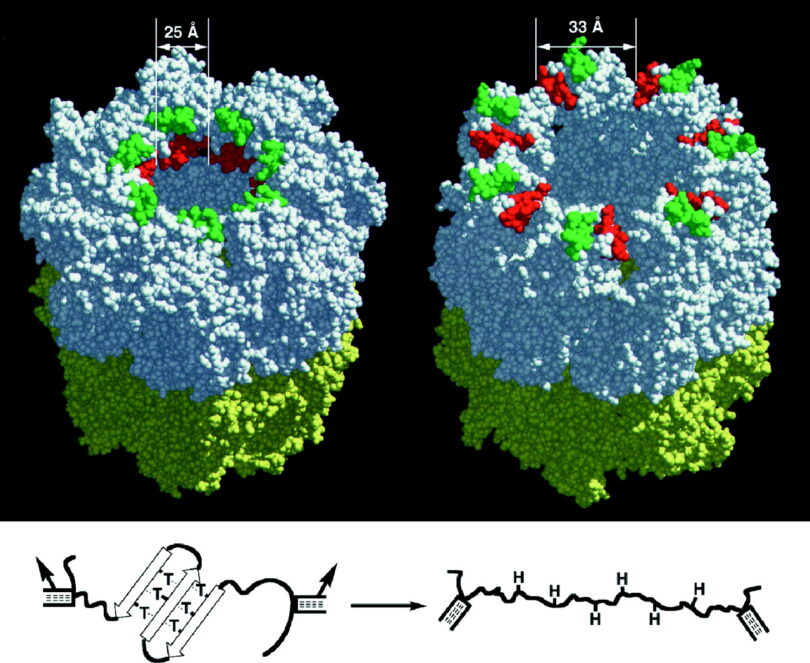
…it is simply amazing to me that this all comes from one very small change, changes in (a single) amino acid…has big effects on all of the other systems in the body that the red cells pass through.-4
Sickle cell disease is a hereditary…caused by mutations in one of the genes that encode the hemoglobin protein…The mutation causes the red blood cells to take on an unusual sickle shape. Individuals affected by sickle cell disease are chronically anemic and experience significant damage to their heart, lungs, and kidneys.”-4
Summary: Clearly, not dying of Malaria is a benefit! Perhaps many millions have survived this horrible disease due to this mutation. However, the mutation caused damage to the red blood cells, which inadvertently provided this benefit. Sounds like an excellent example for universal common descent evolution (UCD)? Well, it is not. Let’s first recognize (as noted in the quote above) that natural selection cannot, contrary to expectations, remove “bad” genetic consequences. This is important to keep in mind when concerning virtually every known mutation directly associated with disease or death. Supposed “good” mutations are exceedingly rare–if they exist at all! If UCD is happening, it must have used an unknown mechanism to clean up all the bad genetic mistakes. However, like those supposed for UCD, any such mechanism remains at large and never observed. These negative genetic consequences due to copy error mutations are called endemic. Endemic means they cannot be reversed in the gene pool and is pervasive in regions or specific populations. Sickle cell was caused by a mutation in the gene that folds for red blood cells that also became part of gametes in the reproduction system of humans. This mutation typically caused round-shaped red blood cells to emerge as deformed sickle-shapes. Because round-shaped red blood cells were the food source for the parasite that causes Malaria, the deformed red blood cells were now effectively invisible to the parasite. Therefore, this mutation caused rigid, sticky, and misshapen (deformed) red blood cells, which counter-intuitively prevented Malaria and saved many lives. However, calling this a “beneficial” mutation does require a strong prescription for rose-colored glasses. This mutation is perhaps the best example of copy errors rendering benefit. However, this was a massive genetic cost of disease, suffering, illness, and death. Like all the other supposed beneficial mutations, the resume is quite ugly, but sometimes even broken things can benefit: Even a broken clock is correct twice every day.
1-https://bigthink.com/surprising-science/evolution-is-still-happening-beneficial-mutations-in-humans/
2- Complications & Damage – Sickle Cell Speaks ;
3- https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/syc-20355876
4-https://www.genome.gov/genetics-glossary/Sickle-Cell-Disease