A strong data-based argument for universal common ancestry of life is revealed when creationists accept the known genetic similarities of house cats and tigers with 95.6% similarity (both from the same cat “kind”) but then reject similar ratios of genetic similarities between chimpanzees and humans (we are the same “kind” as chimpanzees?).
This argument for universal common ancestry evolution appears irrefutable.
However, this inference appears to be so strong due to many of the presumptions made within the comparison and how that comparison is computed. These are not completed universally. Therefore, what we conclude about what genetic similarity reveals can only be fully understood if a more complete understanding of genetics is properly illuminated. This article shall attempt to shine such light upon these topics. We will expose how such claims of universal common ancestry based on genetic similarity are false and exaggerated conclusions.
How genetically similar are chimpanzees and humans?
We have all read that humans and chimpanzees are 99% genetically similar. However, in recent studies, calculations of genetic similarity are found to be between 86.7%1 to 95%2. Other calculations without significant edits are as low as 70%.3
The differences are based on how and what is actually compared.
Any claimed genetic similarity between humans and chimpanzees reported above 95% is erroneous and based on early mapping studies from 2005. All these estimates have been falling as more understanding and analysis of the genetic data is computed. Various reports differ directly due to variable computational techniques between labs.
Also, it is very difficult to accommodate the emerging knowledge base of epigenetic expressions, dormant, neutral, non-coding RNA, and many other vastly unknown variables behind genetic protein-coding. Many of these have been vaguely understood for decades, while many others are just now beginning to be unraveled, even at the most foundational levels. Even more, it remains completely unknown. All these variables and unknowns amount to, at best, an immense difficulty in rendering a clear comparison.
What is “similar” in the genetic similarity exactly?
Genetic similarity compares a very small portion of the DNA sequence totaling a volume of 3.2 billion base pairs (in humans) of specified information programming digital letters.
Only about 1.2% of this total volume of DNA is used for comparisons in a region called The Genome.
This represents 34.8 million genetic letter sequences. When genetic similarities are made, only these protein assembly regions are compared. This tiny region is called The Genome. For genetic similarity comparisons, the balance of the vast 98.8% of the volume is simply considered “genetically irrelevant” and ignored. Despite significant evidence to the contrary, many consider much of the DNA molecule to be “junk DNA.”
Junk DNA?
From a naturalistic perspective, vast molecule segments that are either not yet understood or unknown are often called “junk” DNA. Because there are many more unknown regions within the DNA molecule, initially, they tossed over 98% of it out as “junk”!
Initially, all the remaining 98.8% of the DNA volume outside the protein-synthesizing genome was thought to be “junk DNA.”
Why such a quick presumption that most DNA is “junk”? Under the presumption that universal common descent evolution is correct, it made sense that perhaps these vast regions were garbage or past transmutational relics of a blind chance of trial and error. A junkyard, for now, unused genetic information. Of course, these claims have been soundly debunked. However, like many later recanted messages and click bait of naturalism, these terms continue to be repeated despite empirical evidence the message is completely wrong.
…there used to be an older and derogatory term called junk DNA, which, thankfully, doesn’t (shouldn’t!) get used these days much longer. So really, the thing to keep in mind here that human genome is a vast, vast expanse of nucleotides, 3.3 billion…”
NIH Shurjo K. Sen, Ph.D. Program Director, Division of Genome Sciences, July 2022, Paranthesis and bold are mine.
What does the genome do?
The Genome is a vastly significant and important region of DNA because, despite its tiny 1.2% volume, in humans, it uses these approximately 34.8 million base pairs to provide programming blueprints to the cell.
Genome instructions provide the precise order to arrange amino acids into protein chains which are ultimately folded into three-dimensional structures which form all living material for all life.
Note: As we discussed, geneticists are yet illuminating epigenetic expressions outside the genome, which can also directly cause a variance in protein structures.
Chimpanzees appear similar in many ways to humans anatomically. Therefore, it is not surprising to find that they are also our closest match in the genome that makes these protein structures.
Both creatures live on land, have fingers, legs, arms, lungs to breathe air, hearts to pump blood, and eyes to see. However, we also have many differences, including skull shapes, jaws, necks, spines, limb proportions, pelvis, hands, feet, and soft tissue arrangements such as everted lips, nasal bridge, whites of eyes, and more. Most notably, our brain biochemistry is radically different. Perhaps the most profound differences come in language, art, music, mathematics, technology, philosophy, animal husbandry, agriculture, and moral and spiritual capacity. Also, perhaps surprisingly to the naturalist, the chimpanzee genome is about 12% larger than humans.4 More on genome size distribution problems later.
The total volume of the DNA molecule is known to be more than 80% fully functional
From an intelligent design perspective, we believe most likely all the DNA sequences will eventually be found to be fully functional. Today it has already increased, in some estimates, to be more than 80% functional.5 As we stated before, many non-coding DNA sequences are known to serve functional roles, including the regulation of gene expression. It has been shown that the exact base pair sequences can somehow cause different protein expressions while using even identical genetic coding sequences.6
This means the genome alone does not reveal the depth of the construction of any living organism.
“Epigenetic processes, including DNA methylation, histone modification, and various RNA-mediated processes, are thought to influence gene expression chiefly at the level of transcription.”
Gibney, E., Nolan, C. Epigenetics and gene expression. Heredity 105, 4–13 (2010). https://doi.org/10.1038/hdy.2010.54
Silent (or sometimes unexpressed) RNA sequences stored in DNA can become activated only when very specific circumstances occur. This greatly weakens the assumptions made about the genome similarity between living organisms. It appears that these vast informational strings outside the genome, even outside the DNA molecule itself, serve as a vast library for variety used to ensure life’s survival during highly specific conditions. These discoveries leave very little room to argue why we should not expect to eventually discover that all DNA information will be eventually discovered to be fully functional.
“…over the last decade or so…we are only just starting to realize that there are an immense number of ways in which what we think of as non-coding (outside the genome) actually might just have a more subtle way of passing its information along. So it may not code in the classical protein-coding sense. But there is a ton of information crucial in many, many ways that is hidden in this part of the genome.
Shurjo K. Sen, Ph.D. Program Director, NIH Division of Genome Sciences; “Non-Coding DNA” July 2022-4
Humans and Chimp chromosomes counts are different?
The human genome also consists of 46 chromosomes or 23 pairs, while chimpanzees have 48 chromosomes or 24 pairs.7 A chromosome can be thought of as the library of programming information passed from parents to their offspring that provides the blueprints, from embryo to death, for the assembly of every material structure such as bone, blood cell, enzyme, organ, and hair. At conception, these pairs split; half come from the father and half from the mother (although mitochondria come from the mother only). Together these form new chromosomal pairs (new DNA) for the offspring.
If these chromosome pairs do not match, the species could never have reproduced together without dysfunction or disease.
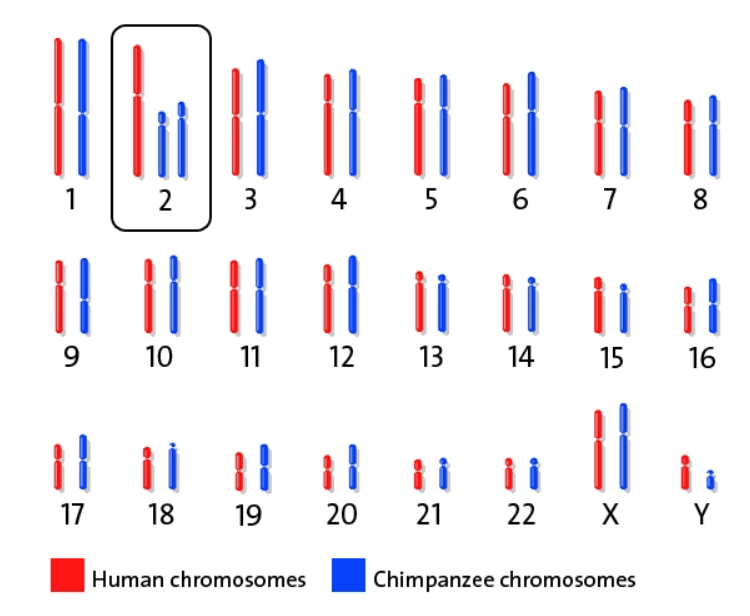
Differing chromosomal pair counts are ignored or downplayed by naturalists when discussing genome similarities despite the ramifications of this reality being immense.
THE SAVE: By begging the question, naturalists presume that two human chromosomes somehow fused together in the distant past. This just-so narrative maintains the theory of universal common ancestry between humans and chimpanzees. Naturalists neglect to mention that no one has ever observed such phenomena. They ignore how such a fusion could have successfully passed to offspring because, as far as we empirically know, such a transmutation would require both the male and the female to simultaneously possess fused chromosomal pairs. This genetic paradox requires a suspension of logic, and a presumption of massive good fortune measured well outside any reasonable mathematical likelihood.
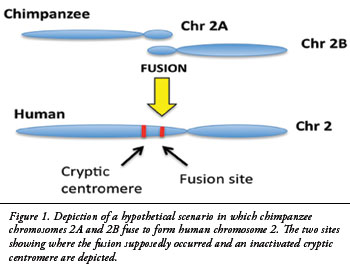
Modern theories used to save this vastly unlikely and yet never observed concept of chromosomal recombination admits that “fusion must have involved loss and rearrangement of part of the genetic material of the two originally separate chromosomes in the ancestors we have in common with the large apes…this rearrangement of chromosomes goes a long way back in time: estimates using various methods date this from 0.75 to 4.5 million years ago.” (Note: this is quite a range!)
Meyer et al. 2012 A high-coverage genome sequence from an archaic Denisovan individual. Science, 338:222-226.; K. H. Miga. 2016. Chromosome-specific Centromere sequences provide an estímate of the Ancestral Chromosome 2 Fusion event in Hominin Genome.Journ. of Heredity. 1-8. Doi:10.1093/jhered/esw039. https://www.bbvaopenmind.com/en/science/bioscience/the-origin-of-the-human-species-a-chromosome-fusion/ Bold in the original. Paranthesis and italics are mine.
Chromosomes are thread-like structures located inside the nucleus of animal and plant cells. Each chromosome is made of protein and a single molecule of deoxyribonucleic acid (DNA). Passed from parents to offspring, DNA contains the specific instructions that make each type of living creature unique… Changes in the number or structure of chromosomes in new cells may lead to serious problems. For example, in humans, one type of leukemia and some other cancers are caused by defective chromosomes…”
https://www.genome.gov/about-genomics/fact-sheets/Chromosomes-Fact-Sheet, bold is mine.
Orphan Genes
Orphan genes are a serious challenge to universal common ancestry because these genes are unique only to that life form. Naturalists cannot point to genetic similarity as an explanation because the gene sequences are exclusive.

THE SAVE: Universal common ancestry provides its save for these unique genes within the name itself: orphan genes. This term provides the assumption, again begging the question, that these unique sequences must have been “orphaned” by preexisting sequences that no longer exist.
It is highly suspect because an orphan is an offspring its parents abandoned. However, orphans are still biologically derived from these parents. However, the term orphan genes refer to the numerous base pair genetic sequences within the genome that are unique and found only in that particular creature (roughly at the taxonomic family level), meaning no genetic similarity.
“A recent report found at least 1,307 different genes (“orphan genes”) between humans and chimpanzees” with “634 in humans and 780 in chimpanzees.”.
Ruiz-Orera, J. et al. 2015. Origins of De Novo Genes in Human and Chimpanzee. PLoS Genetics. 11 (12): e1005721.
Orphan genes are distinct programming code that makes the creature unique and distinct from other life forms.
These genes provided functional biological processes, trait expressions, and highly specialized adaptations. In addition, the advent of epigenetic discoveries complicates this problem further as the combination of other RNA transcript expressions with complex coding and non-coding regions is also largely ignored.
“For the past 20 years scientists have puzzled over a strange-yet-ubiquitous genomic phenomenon; in every genome there are sets of genes which are unique to that particular species i.e. lacking homologues [similar counterparts] in any other species.”
Ruiz-Orera, J. et al. 2015. Origins of De Novo Genes in Human and Chimpanzee. PLoS Genetics. 11 (12): e1005721. Bold is mine.
This disconnect logically infers that although various species might have similar anatomical construction to survive similar environments, differing codes without a clear connection back to a presumed common ancestor falsify this rationale and the theory of universal common descent.
“Orphan genes are “the hard problem” for evolutionary genomics. Because we can’t find other genes similar to them in other species, we can’t build family trees for them. We cannot hypothesize their gradual evolution; instead, they seem to appear out of nowhere. Various attempts have been made at explaining their origins but the problem remains unsolved.”
“The evolutionary mystery of orphan genes-Every newly sequenced genome contains genes with no traceable evolutionary descent – the ash genome was no exception
Richard Buggs. Professor, Queen Mary University of London; Dec, 2016. https://ecoevocommunity.nature.com/posts/14227, Bold is mine.
Genetics employ a digital language
DNA is a language similar to a digital written or computer programming code. DNA uses a quaternary system (four characters derived from A, C, G, and T, representing the four nucleotide bases of a DNA strand — adenine, cytosine, guanine, thymine) that use three letters to call for specific amino acids. The genome and regions outside the genome together provide the blueprints used by the nano-factory of the living cell to assemble twenty different (amino acid) “letters” into protein chains. Charts can make it appear complicated, but codons call for amino acid “letters,” which are assembled into protein chains just as letters in a digital language are arranged into words and paragraphs.
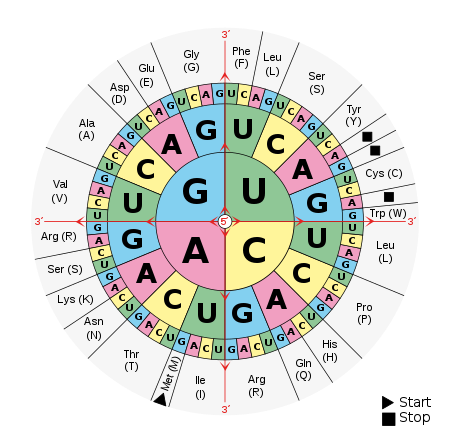
It has been discovered that this language uses punctuation, syntax, letters, phrases, and even replicated chapters assembled in a specified order to form what we call genes. DNA goes to great lengths to avoid errors (spontaneous mutations) during replication. The molecule effectively prohibits transcriptional and replicational errors to as high as the eleventh decimal. Complete with onboard error-correction, self-replication, self-modification, self-diagnostic, self-repair, and self-reproduction capacities, the molecule deploys enzymes that run the length of the molecule that not only identify copy errors but also correct them as well! This is astounding to consider that a tiny enzyme could carry that much information and sophistication! Arguably this enzyme is the most complex and sophisticated phenomenon in the universe!
Together these mighty enzymes and many other read-write protections provide the molecule an astounding exceedingly tiny copy error rate (mutation rate) of one occurring every .000000000001 times. Or one copy error for every ten billion letters.
Whether naturalists agree that DNA is a digital language or not, the reality of this massively complex and rigidly ruled programming code is undeniably profound and sophisticated beyond our understanding. Certainly, as naturalists often do, arguing that the Universal Genetic Code emerged by chemical affinities, chance, time, predestination, and Natural Selection seems to defy common sense and even our own eyes.
Genetic Similarity is merely comparing amino acid sequences or “letters.”
Comparing genetic similarities to a written language helps illustrate some problems. For example, “I woke up at noon today with a headache” compared to “I woke up at noon today without a headache” is 93% identical but means two different things.
The first statement has 39 characters (including spaces) that would call for three codons for each letter or 117 (G, T, C, A). The second phrase adds “out” to a “headache,” so it has three additional characters or 42 letters or 126 characters. That is a ratio of 117 / 126 = or about a 93% identical code similarity. This similarity would become even greater if the protein chain was much longer and the average gene was more than 350 amino acids long.
To add fuel to this problem, computational methods would realign the terms and ignore the “out” part, resulting in a 100% code similarity. I know this sounds incorrect, but it isn’t.
The significance of written language is not found in similar letter sequences but within the composition of those written sequences that might convey different meanings.
In the first case, there was a headache; in the second, it was without a headache. This is why one cannot compare written work merely by the sequence of letters, especially using realignment techniques at tabulation. The idea is preposterous, yet this is precisely how genetic similarities might be calculated. More on this later.
To illustrate this a bit further, we might compare the letters of various novels. Especially if we did not fully understand the language, we would look for textual similarities. Assuming the books were written in the same digital language, we would find that the same digital letters were used, there were many similar phrases, identical sequences, and similar formats. We will observe that it appears that the same rules (or syntax) were consistently used to separate intended meaning from potential gibberish. However, we expect that these language similarities do not infer that the two written novels have the same meaning. Also, as we have illustrated, a few digital characters (letters or amino acids) can radically change the meaning or the intended purpose altogether.
With all that said, we also must not neglect to understand that although The Universal Genetic Code is highly similar to a written digital language, it is not a language that is linear. Genetics form proteins that fold into three-dimensional shapes. Even a single letter or punctuation can significantly affect the gene.
“(Another) problem is that we tend to think of DNA sequence as a human-written language, in standard linear format similar to the English 26 letter alphabet. Such reasoning evaluates differences as if one would line up parallel written texts. Two books written by humans that are 98% similar are essentially the same book. Evolutionists often use this analogy, but it is completely inappropriate. The DNA four-letter alphabet code that designates twenty different amino acids by codons (triplet bases of specific sequences) considers only the small fraction of the genome that actually codes for protein.
https://creation.com/human-chimp-dna-similarity-literature
The point here is much of the protein expressions come from outside the genome. In other words, if the genome was a large written book for protein-coding, outside the genome, there are at least eight more books of similar size that are used to express the purpose of the genes. Considering how little we yet understand about the DNA molecule is astounding.
Today, similar sequences within normal genes can be used to find specific abnormalities (caused by spontaneous mutations) that can “predict an individual’s risk for physical and mental disease based on their genetic profile.”3 However, when comparing similarities between various living organisms, no baseline has been universally established; each lab uses different methodologies and realignment techniques .3 This opens a great latitude for researchers to cherry-pick genetic data.4
Genetic material used for comparisons are cherry picked
When genetic similarity percentages are computed, “For exome or genome sequencing, potentially millions of variants are identified that differ…(only) the “reference sequence” (are) used for
comparison.” This is used when looking for genetic mutational abnormalities between parents, offspring, and living organisms.
Nonetheless, the idea that sequences are factually highly similar is grossly exaggerated because millions of points of genetic informational code, which are not yet fully understood or even considered “junk,” are routinely excluded. These vast regions are considered to be genetically insignificant. While such exclusions make sense when comparing sequences to identify genetic abnormalities (degradative mutations) of offspring from parents, such exclusions between creatures become very difficult to assess and open to highly subjective alignments and deletions.
All the alignments and similarity calculations are based on a begging-the-question fallacy that preassumes that common ancestry evolution is correct, which is why we see assume common ancestry in genetics. The material cause cannot be the same as the material effect. This logical fallacy is akin to saying the wind is blowing because it is windy. There is no information in that statement–it is null.

(Studies, most often) report the ‘best of the best’ data—a form of dogma-driven bioinformatic cherry picking. For example, only the “protein-coding gene sequences of preselected highly similar DNA are used”6 guaranteeing high levels of similarity. Perhaps the most widely cited paper that reported the initial 5x rough draft of the chimpanzee genome assembly is the most errantly cited…”the overall actual DNA similarity compared to the human genome… (from 95% to only) about 70%”.6
The genetic code is designed to minimize deleterious mutations
Devastating to the notion of life arising by natural processes due to chemical affinities, atomic characteristics, and natural laws are revealed in how the arrangement for coding within the DNA language works within its construction to counteract copy error mutations (which are undeniably deleterious).
DNA is a long string of “nucleotides” (in humans is 3.2 billion base pairs in length) comprised of four different chemical structures referred to by the letters A, U, C, & G. The specific order in which these nucleotides are arranged determines what type of protein will be made in the living cell. U and C are in pairs, as are A and G.
Proteins are vast strings of amino acids averaging about 350 in length, with the longest being the super protein chain called titin at over 34,500! There are 20 different types of amino acids used throughout all life. Groupings of three nucleotides are called codons. Each codon calls for one specific and unique amino acid.
The genetic code, charted below reveals an important revelation, if you had DNA with nucleotides in the order of A,U,G, U,C,U, C,A,U, U,A,A, then this would render a protein with the amino acids, methionine, serine, histidine, and “stop“.
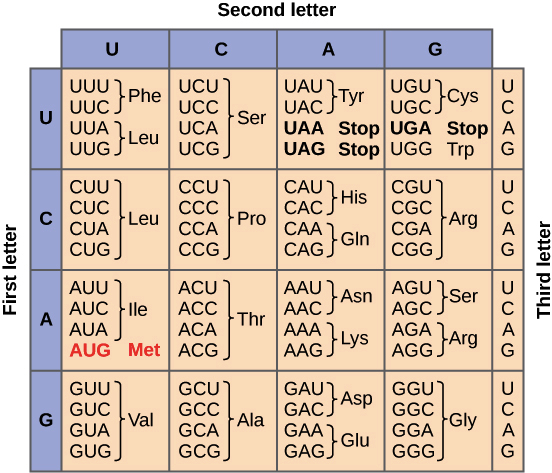
If the DNA sequence is incorrectly translated during replication, say U to a C, or a C to a U, or an A to a G, or a G to an A (remember these are in base pairs), then in either the first or third position of the codon, the result is almost exclusively the same; We end up with the same amino acid (or one with extremely similar chemical properties) every time! This is extraordinary!
This code isn’t haphazardly slopped together by some random biochemical accident. It is revealed as being perfectly optimized!
“Here’s the takeaway: the genetic code was created/established/locked into place before evolution could begin to occur, so evolution may not be used as an explanatory device. Before evolution may ensue, you must first create a system that would allow for the replication of DNA, so that the very first living cell could duplicate itself, and pass on its DNA along…This replication system requires the genetic code to already be in place as it relies on proteins to work. The genetic code was first created before the replication system, but it seemingly knew beforehand the errors that the replication system would be prone to and created itself in accordance with that foreknowledge.”
Deane, Corey; “What is the most rational evidence, if there is any, of the existence of God”? Quora post July, 2022 https://qr.ae/pvMJ1Z
Genome sizes defy evolutionary expectations
Diagrams in many textbooks depict humans as being the most “evolved” and therefore are found at the very top of various charts: at the top of tree branches in Darwin’s “tree of life” and at the top of many other genetic diagrams. Such placement is assumed to result from evolutionary mechanisms continuously building better and better life forms over time. As Darwin put it, these mechanisms (spontaneous copy error mutations) have been “adding up the good” for millions of years. Therefore, based on such reasoning, humans should have the largest genome size. But we do not, and such depictions are deceptive.
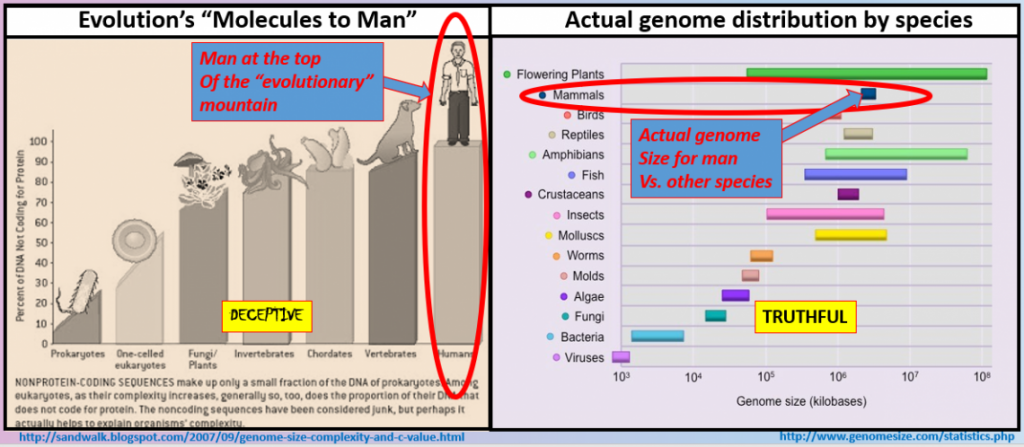
The honor of the largest genome size belongs, shockingly to naturalists, to flowering plants.
The internet, textbooks, universities, and most all evolutionary teaching shows the famous ‘simple’ life forms becoming more complex over millions of generations in a web of transmutation. We all know the classic monkeys to man line up.

However, these arrangements are imaginary and not based on the evidence but on the narrative. Scientific discovery based on genome sizes within the variety of life challenges this dogma directly. Genome sizes indicate that what evolutionary mechanisms may have expected to find in the ‘simple’ to ‘complex’ narrative simply does not pan out from the genetic and physiological evidence. While such evidence defies the narrative of evolution, these various genome distributions can be explained within a creationist perspective of intelligent design. God provided the necessary design and programming for each unique living thing based on its intended function.
Mutations are bad
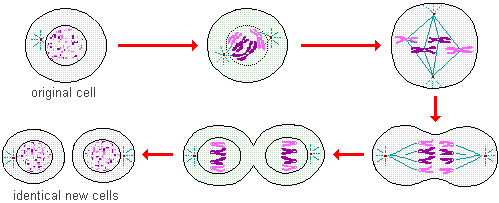
Before we get to why mutations are bad, we need to review a few definitions. All living things are composed of Cells, each human has many trillions of cells. Somatic is a term in biology which refers to physiology or specifically that which pertains ultimately to the cells within the physical body. Germline is a term in biology that ultimately refers to the process of how cells progress in a series; specifically, here, we are referring to the sequences in heredity. Heredity is genetically passing traits from one generation to another or reproduction. Heredity, for our purposes here, is the same thing as natural selection. At conception, germline cells come from the parent sex cells (sperm and ovum), delivering their respective chromosomes to form a unique new offspring. The traits that are passed come from these combined chromosomes (bundles of DNA) from the parents in specialized regions called Alleles. Alleles are gene variants that provide options for a vast variety at conception. The frequency that similar traits emerge, such as brown eyes compared to blue eyes, are called Allele Frequencies. These frequencies are commonly known as dominant (brown eyes) or recessive (blue eyes) traits.

Mutations, properly defined as spontaneous copy errors, which occur rarely, do accumulate nonetheless. These copy errors change the genetic sequence by substitution, deletion (which causes a frameshift to the sequence), inversion, or insertion (called a point mutation).
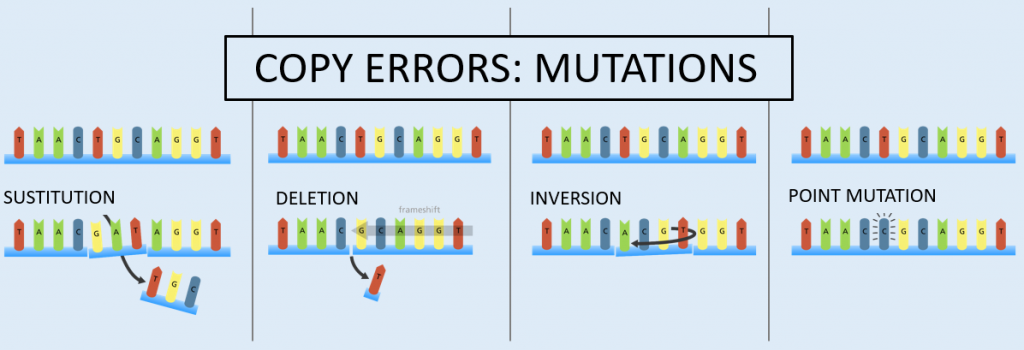
These mutation types impact both somatic (body) cell genetic sequences (somatic mutations) and the genetic sequences within the sex cells (germline mutations). Somatic mutations occur in body cells and are not transferred to offspring. Germline mutations occur within the sex cells.
This is important: Heredity does NOT operate by copying error mutations to the germline as it functions by allele gene variants provided by the parents to the offspring. This is where “bait and switch” habitually is used by evolution popularizers, interchangeably misusing the terms heredity and mutation despite these having opposite meanings.
Heredity refers to function, and mutation refers to random copy errors to that function.
Most definitions avoid stating the need for beneficial mutations altogether despite the necessity as a mechanism for universal common ancestry (modification by beneficial mutation is also called the “modern synthesis”).
Let’s examine the most common definitions and find the root of the modern synthesis:
“…change in the heritable characteristics of biological populations over successive generations.” Heredity.
“…characteristics are the expressions of genes passed on from parent to offspring during reproduction.” Heredity.
“…a gradual process of change and development.” Heredity.
“…descent with modification.” Heredity.
“…any net directional change or any cumulative change in the characteristics of organisms or populations over many generations.” Heredity by allele frequencies.
“Different characteristics tend to exist within any given population due to mutation, genetic recombination, and other sources of genetic variation.” Ah! There it is! Different characteristics because of mutation! Plus, “other sources of genetic variation” are Heredity.
No one is arguing this fact. Somatic mutations are the primary cause of most cancers, tumors, and diseases. Even naturalists are not expecting somatic mutations to derive anything beneficial. Most will admit that these mutations are very harmful.
This is supposed as the mechanism that “adds up” beneficial copy errors over eons of time, passed to offspring, and then locked in by heredity (“natural selection”). Heredity by allele frequencies does not provide the mechanism required for universal common ancestry evolution–only mutation does. Therefore, if germline mutations empirically can provide fitness gains, they can plausibly infer a pathway to theorize universal common ancestry. If germline mutations are bad, then the theory is at best weakened and at worst falsified.
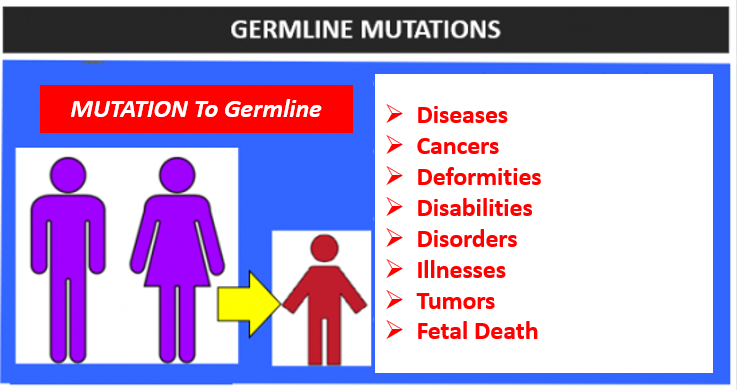
Germline cells, like somatic cells, are affected by mutations. Somatic mutations are all objectively degradative but are germline mutations good? No.
Germline mutations have seen many billions of dollars spent on research. This is not because these mutations are good, but because they are very harmful.
Offspring that survive the womb with germline mutations are affected with countless diseases, cancers, propensities toward cancer, deformities, and disabilities like Autism, and illnesses, with some even leading to premature death.
The naturalist will argue that there are “many” good mutations. These fall into four primary categories: (a) ancient, (b) anecdotal, (c) heredity, or (d) actual lab-verified germline mutations.
(a) Ancient claims (based on begging the question) speculate that our genes today “must-have” mutated from thousands or even millions of years ago. A couple of examples of these include lactose tolerance and blue eyes. Seeing no fully sequenced DNA molecule exists from thousands (or millions) of years ago, these are only speculations.
(b) Anecdotal claims lack evidence or are not objectively validated. As an example, tetrachromatic vision was claimed by one individual (woman) out of twenty-four studied. This claim of superhuman vision is merely anecdotal at best, and no one can measure IF the trait supplies more color vision than is measurable. Essentially anyone with the mutation could claim the ability, and no lab measurement could prove or disprove the claim.
(c) Heredity is usually the cause behind most claims that involve verifiable and measurable traits changes in life. As we have discussed extensively, these varieties are provided by allele frequencies that have nothing to do with mutations. Examples include elephant ivory tusk production decreases or wisdom teeth count differences in humans.
(d) Lab-verified mutations confer a benefit or fitness gain. Yes- these at least subjectively exist (I will elaborate on why below). Of these lab-verified mutations, what caused gene sequence modifications AND conferred a fitness gain are the very “beneficial” mutations the entire theory of universal common ancestry hangs upon.
Lab-verified “fitness gains” are subjective
All lab-verified fitness gains are likely (all) subjective. Let me explain first by analogy: Let’s imagine that we discovered a mutation that blocked calorie uptake significantly. If you are overweight, you would most likely see this as an awesome benefit! However, this hypothetical mutation could prove deadly if you are impoverished or even of normal weight.
If we compare our analogy to a real-life lab-verified mutation coined the “good cholesterol mutation” (aka Apo-1 Milano mutation) that has a claim to fame of blocking “bad” cholesterol within the liver. Again, just as in our hypothetical example, if you are overweight or do not eat a balanced diet, you are likely to consider this a beneficial mutation, but others worldwide can develop chronic diseases. Therefore, this illustrates how this is a subjective “benefit.”
As a final analogy: It is like having the air conditioning in your car broken by a leak in the system, but instead of focusing on the leak, we focus on aspects that benefit. We find no air conditioning means better gas mileage. However, did we JUST “gain” better gas mileage? Subjectively yes, but objectively NO. Mutations break preexisting gene functions, which can, very rarely, counterintuitively provide a subjective fitness gain.
The body, composed of trillions of living cells, is a dynamically complex and interdependent system of proteins and living apparatuses. No single change ever happens in a bubble. Any change to any protein or apparatus WILL impact many systems. As evidence of this reality, we find that even the world’s best medicines also carry adverse side effects. Likewise, ANY mutation to ANY gene confers a measurable fitness change and will also confer side effects. This is not an opinion, it is a verifiable fact. I have listed all the supposed and subjective “good” mutations here. Anytime someone proclaims a new specific “good” mutation, add the term “side effects” to your Google search and learn how subjective the claim is.
Perhaps the best example of a “good” lab-verified germline mutation that conferred a fitness gain is The Sickle Cell mutation.
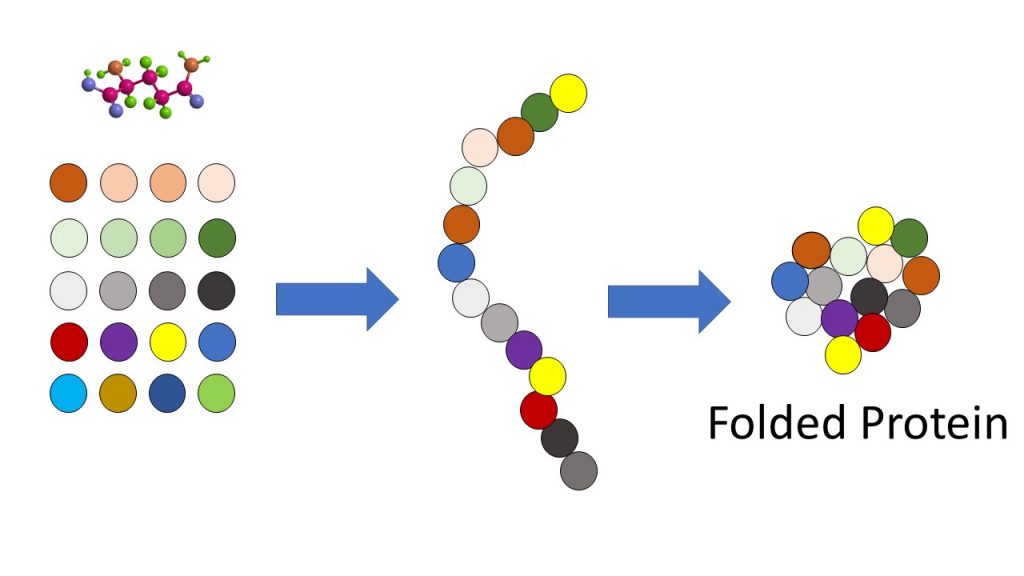
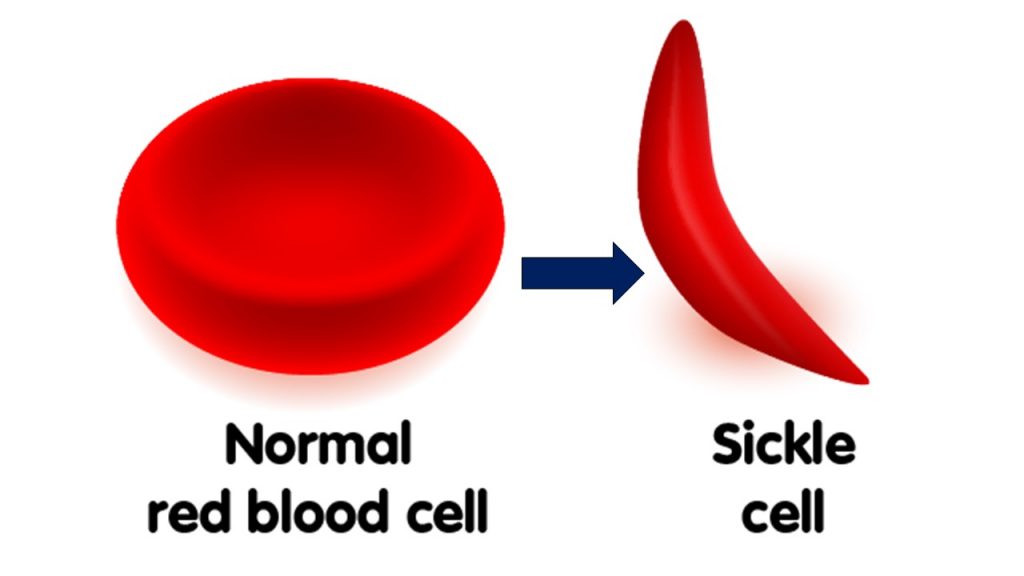
Any non-silent mutation can cause a devastating misfolding of a protein chain. This “problem” for evolution is called Levinthal’s Paradox. Here, with the Sickel Cell mutation, the proteins folded wrong and deformed the red blood cells into sickle shape (thus the name). This mutation, counterintuitively and serendipitously, provided an inordinately rare “benefit” by causing those affected with the trait to “gain” the ability to survive Malaria. This was a very fortuitous result. Here is proof of beneficial mutations…right? Not exactly.
Because the pathogen (a parasite that eats round blood cells and causes the deadly disease Malaria) could no longer recognize the sickle-shaped red blood cells as food, it died off. This resulted in millions surviving Malaria. However, Sickel Cell is still an endemic (can never be reversed) genetic disorder that causes massive health problems for the carriers and their offspring.
And this is the poster child for “good” germline mutations!
The balance of (all) other lab-verified germline mutations is listed within the tens of thousands of pages of genetic research that documents diseases, cancers, deformities, tumors, illnesses, and more devastation. The same germline mutations directly cause all these ailments Neo-Darwinian evolutionists point to for all the goodness and variety in nature. There is a clear disconnect here!
The differences between somatic and germline “mutations” are the most frequently used bait and switch by proponents of evolution using allele function disguised as a mutation.
Genetic researchers have been vigorously and diligently searching for “beneficial” mutations in germlines. The effects of such beneficial mutations must be there somewhere as they are presupposed to explain all the variety and order within life as we observe it today. However, again, without evidence, this logic is merely begging the question. Exactly where is the vast evidence of transmutation and such beneficial mutations? This is a big question and a massive “problem” for the theory.
In reviewing numerous reputable sites, it takes very little time to discover that beneficial mutations actually are “rare” and “scarcely occur” .
Desai MM, Fisher DS. Beneficial mutation selection balance and the effect of linkage on positive selection. Genetics. 2007 Jul;176(3):1759-98. doi: 10.1534/genetics.106.067678. Epub 2007 May 4. PMID: 17483432; PMCID: PMC1931526. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1931526/
When the mountains of data are tabulated, the experimentation is reviewed, and research is completed, ANY mutation (as a copy error) to the germline is found degradative or, at best, a subjective “gain.” These mutations have never been shown to build one new gene. Empirically germline mutation can only break fully functional programming code. These mutations are not “evolving” new physiological features (sorry, no wings for you!).
Germline or Somatic mutations are not the heroes, they are the enemy.

Mutations are objectively hazardous killers that break preexisting gene sequences that render dysfunction to death. These facts are undesirable for naturalists to accept, but they are undeniable. Together empirical evidence of mutations deals a death blow to the theory of universal common ancestry by falsifying its mechanism: mutation.
“We have studied and analyzed the whole genome, and our analyses of mutations that are affecting cancer genes have enabled us to genetically explain 95 percent of the cancer occurrences we have studied by means of mutations.”
Dr. Joachim Weischenfeldt; http://citechdaily.com/massive-genetic-map-of-cancer-mutations-cataloged-available-to-doctors-and-researchers-worldwide/
“Benefical” mutations are exceedingly rare (if they exist at all)
Only germline mutations can provide the mechanism for transmutation or universal common ancestry of life. These mutations must render new gene sequences that confer benefit. These copy errors must be passed to offspring and confer “benefit” or a fitness gain to the organism.
Biologist Douglas Axe studied this question directly. He examined the likelihood of random copy errors in preexisting genetic sequences conferring a benefit. In other words, did the supposed mechanism behind universal common ancestry evolution empirically exist?
Axe used a profoundly simple protein (gene) of a modest 150 amino acid length (average is over 350). He constructed a model that estimated the odds of a single new protein fold being introduced within the known mutation rates as observed empirically in the lab. He reasoned that the simplest known protein folds to render a new gene is called a “hook.” A hook consists of only two amino acid alterations (in most cases). The results of the study found that the likelihood of such an occurrence ever happening, even once, was vanishingly low.
The problem was mutations are much more likely to destroy a gene than help it. Trying to get two mutations to add a hook proved mathematically absurd.

Based on a chart in Darwin Devolves by Stephen Myers as reproduced from “The new science about DNA that challenges Evolution” by Michael Behe
Axe determined that a random mutation conferring a hook protein fold had a minuscule 1 in 1077 chance of occurring even once.8 That is one followed by seventy-seven zeros! This is a nearly incomprehensible and absurd unlikelihood. To put it into perspective, there are an estimated 2.4 x 1082 atoms in the entire universe!9
However, Axe recognized that there were many opportunities or trials for this random event to have happened considering all the vast number of life the earth has had over its (presumed) 4.5 billion year history. There was one new trial at conception (or replication) of each life form that has ever existed on earth (he included both extant and extinct life). Side note: The experiment presumes life already exists with DNA sequences and the pre-existing gene that can be mutated. How this first life form existed is another matter altogether. The estimate (including yeasts, funguses, and bacteria) was 1044. 8
Axe divided these 1077 / 1044 (exponents cancel out in division 77 – 44 = 33) and calculated the chance of a simple hook protein fold emerging on a preexisting gene sequence (on a 150 amino acid chain) throughout all life that ever existed on earth, of happening only ONCE was a remote 1 in 1033.8
That is a ten trillion, trillion, trillion, million to one chance!
The researcher calibrates molecular Clocks
Molecular Clocks are proposed as a solution to calculate the genetic “distance” of all living organisms. The greater the genetic variance of the DNA code, like a ticking clock, the greater the time distance since the supposed divergence. The greater the genetic differences, the greater the time since the organisms split (diverged).
Within living organisms, there are many proteins (gene sequences) for the researcher to select for their study. Some proteins mutate rapidly, while others are slow, rarely mutating.
Therefore, the protein the scientists select for their model will ultimately drive or calibrate their molecular clock.
Scientific computer models render different results based on the study’s selected protein sequence. Therefore, like Goldilocks and the Three Bears, the researcher selects the presumed protein mutation rate (based on much speculation) to be “just right.” That protein that provides the most “correct” age is based on begging the question that presumes the cause and effect of universal common ancestry is correct.
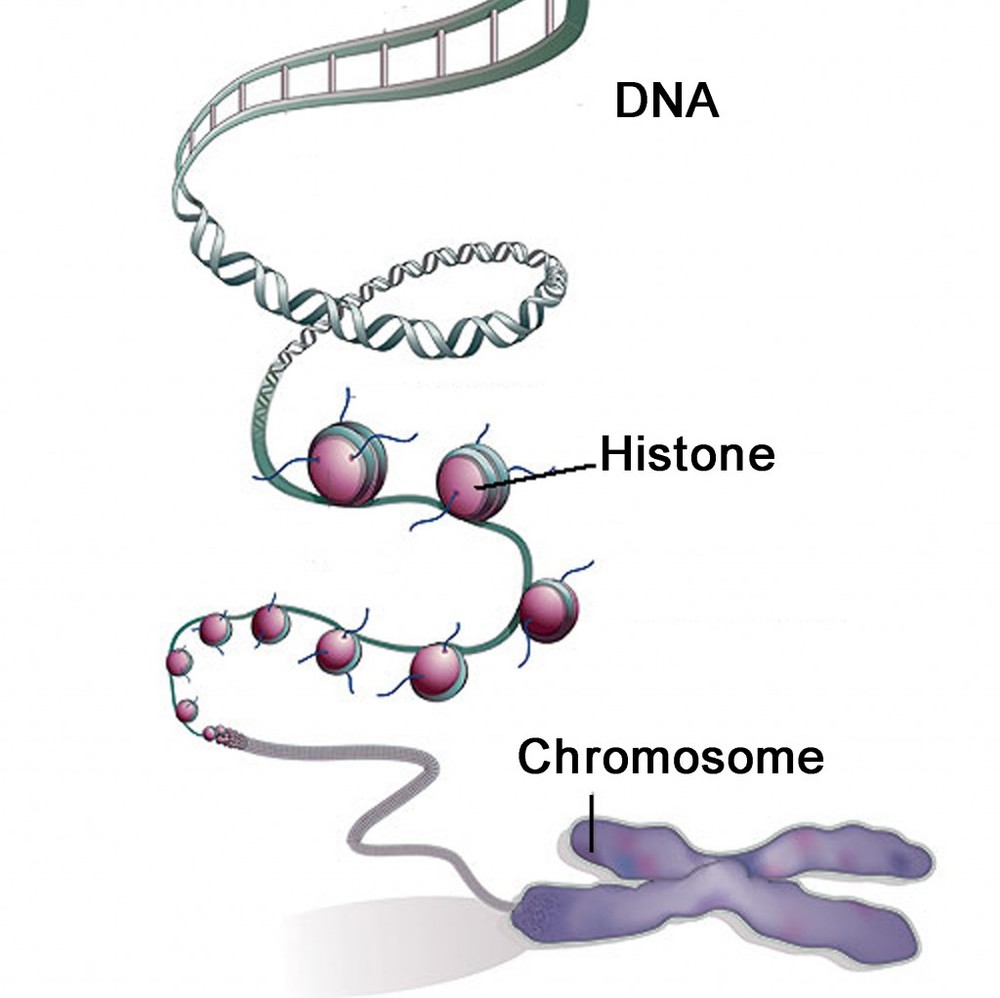
Histones are DNA proteins found in all living organisms, from bacteria to mankind. DNA chains are wrapped in bundles called chromosomes. DNA strands are wrapped around histone proteins. If the researcher selected any universal protein for proper study, it seems the best candidate would have to be histones because all life forms have these within their DNA.
“…small differences between histones yield an extremely recent divergence, contrary to other studies.”
Axe DD. Estimating the prevalence of protein sequences adopting functional enzyme folds. J Mol Biol. 2004 Aug 27;341(5):1295-315. doi: 10.1016/j.jmb.2004.06.058. PMID: 15321723.
However, histones are never used in molecular clock models because they exhibit too few differences (or presumed ancient mutations) between species. Therefore, histones indicate a recent divergence of all species, which, of course, cannot be true based on the presumptions that evolution has been ongoing for billions of years. So the researcher must select a different protein with less similarity (the “just right” amount). The protein must have (presumably) incurred more mutational changes (unverifiable in the distant past), based (again) on a begging-the-question logical fallacy that the differences in modern genetics all happened by mutations in a distant unobservable past despite no such genetic evidence supporting this conclusion today (outside begging the question). Again, the effect of universal common ancestry in genetic differences must have been caused by universal common descent by mutations. Regardless, because different proteins have different variances between species does not mean they transmutated into that condition–it means they are different. Because the researcher can select from many proteins, they can render any desired result. Therefore, using the same assumptions (method) but selecting different proteins, the creationist can have results of life only about 6,000 years old. In contrast, the naturalist can select a different protein and render results of billions of years. This renders the tool of molecular clock models invalid.
“The rate of molecular evolution can vary considerably among different organisms, challenging the concept of the “molecular clock.”10
Reproduction defines “kinds”
Naturalists usually despise the biblical categorization of living things made within the bible called “kinds.” No one has any confusion about the term humankind, but it is arguably a vague and imprecise term. However, our comparison here ultimately contrasts universal common ancestry evolution and the creation account, so we must discuss the term “kind.”
First, naturalists are just fine debating over more than 25 different models for the term “species“11 alone but become unhinged by the term “kind” when used by creationists. Despite recent modifications to the term species, until about the middle of the twentieth century, the meaning of “kind” and “species” was vastly similar as both referred to lifeforms that could successfully reproduce.
Therefore, to clarify, the classification of lifeforms as “kind” means those creatures that can and do successfully reproduce at the origin of life- at the time of creation. With this stated, knowledgeable creationists recognize that the vast diversity of life around us today was ultimately provided by heredity (as gene variants called alleles). During these thousands of years, speciation occurred. Speciation as life forms that once reproduced but, as of today, no longer reproduce. Some of these can render offspring as human-induced hybrids. One example (of many) is artificially impregnating a donkey with zebra sperm and thus rendering a “zonkey.” These animals would not likely reproduce naturally as they live in different environments and have developed different social skills. Still, other life forms can no longer reproduce today.
Speciation, isolated populations, genetic bottlenecking, or specialization are not examples of transmutation (universal common ancestry). These are variance is driven by trait variances provided by preexisting gene variants of DNA called alleles. Allele frequencies confer (oftentimes rapid) variability- not mutations!
The similarity in programming language points to intelligence and design
As we discussed briefly above, genetic similarity makes sense and is not surprising to find within living creatures that share very similar environments. This is even more pronounced in living creatures that appear physiologically very similar, as we observe between humans and chimpanzees. However, based on a theistic and creationist perspective, we can propose a very obvious explanation for genetic similarity. That is intelligent design.

“Common genetic code is a predicted feature of purposefully engineered systems in the genomes of creatures that share the same environment and have similar requirements. It’s just like a computer programmer who uses common pieces of code in different software programs.”
Ruiz-Orera, J. et al. 2015. Origins of De Novo Genes in Human and Chimpanzee. PLoS Genetics. 11 (12): e1005721. Quoted from ICR article: “Genetic Gap Widens Between Humans and Chimps” Jeffery P. Tomkins, PH.D., Jan, 2016.
The only known source we have ever observed for highly specified programming languages we find used in computers or written languages is intelligence derived from a mind. It is not unusual to reuse code for similar functions when computer programmers write code. For example, a computer programmer would not write brand new code from scratch for a calendar, they would plug and play. Similarly, the programming language for life is universal and infers its origin from a mind.
What else are humans genetically similar to?
Undeniably, animals of the same family (same kind) share very similar genetic sequences in the genome (99.6%+ between all humans). Yet, lesser similarities do not defend the theory of universal common ancestry of life when placed under scrutiny. As we have discussed, the calculation methods are highly suspect and subjective, with techniques varying from lab to lab. However, we know a high similarity between chimpanzees and human genomes is real.
But what other living things are humans also genetically similar to?
Bananas!
The National Human Genome Research Institute found that humans and bananas share up to a 60% gene similarity!13

What?
Scientists compared all banana genes to all human genes, counted similar genes, and found that about 60 percent of our genes have a counterpart in the banana genome. Of that 60 percent, about 40 percent were identical when compared to the amino acid sequence of the human protein.”
“Do Humans have the same DNA as bananas?” Jamrozy, Kelly; Medically Reviewed by
Dr. DHarshi Dhingra MD, with a specialization in Pathology.
Here, unlike chimpanzees and human comparisons, the article minimizes the significance of this genetic similarity by pointing out the weaknesses of the genome as we have done here in this article. They point out that the genome is (i) less than 2% of total DNA volume, (ii) both are living things, (iii) both exist in similar environments and (iv) even points out that the balance of the other 98% of the DNA volume holds the key to the answer! Much of the protein expressions, eight times the volume of the genome, come from informational bearing sequences within the DNA molecule but outside the genome!
What does the other 98 percent of our DNA do? Eight percent (as currently understood) determines if genes will be turned on or off. It’s responsible for regulation. The other are of unknown function or they no longer function. Scientists sometimes call this… “junk DNA”. However, as more is revealed about our DNA, scientists are finding that some of the junk DNA are functional,”14
What other living things do we share high genetic similarities with? A mouse 85%, 60% with fruit flies, 80% with cows, and many other genetic similarities with other highly discordant life forms. One, in particular, using the same techniques, finds that the Abyssinian house cat shares a stunning 90% genetic similarity to humans.13
The numbers (in genetic similarity tabulations) can be misleading, though, because much of the shared DNA is “silent” and is not involved in the coding sequence.”14
Conclusion
We have uncovered (1) how the genome size is only about 1.2% of the total volume of DNA and how at least eight times that outside the genome (at least) controls protein expressions. These regions are excluded in almost all genetic similarity computations; (2) We know that chimpanzees and humans have different chromosome counts that prohibit reproduction; (3) We found that despite its catchy name, orphan genes were unique gene sequences found only within that particular creature. There are no transmutational descent explanations that suffice to explain these unique and highly specialized genes; (4) We proposed that the clear ramifications of a sophisticated written programming language complete with punctuation, syntax, and read-write protections were better inferences for intelligent design and not random nature; (5) We found that different labs used many different computational techniques that re-realign, ignore, and tabulate supposed similarities differently making claims exaggerated and unreliable; (6) We discovered that genome sizes throughout very different life forms simply do not make sense within a transmutational model. Why do flowers have the biggest genome?; (7) We empirically know that mutations in the present are exceedingly rare. (8) We also factually know that when mutations manifest, somatic or germline, they are always degradative. These mutations undeniably directly cause countless diseases and even death. Finally, (9) we found that genetic similarity within the genome alone is vastly weak, as revealed by humans having high genetic similarities to bananas and fruit flies.
All these factors must be considered when considering the claims of human and chimpanzee genetic similarity. We must evaluate all the data in a vast sea of supporting and opposing points. Is there a high genetic similarity between humans and chimpanzees? Yes! Especially under the bright lights of scrutiny as we have applied here. However, we have illustrated clearly that even very high genetic similarities in the genome do not convey the entire life story.
Genetic similarity cannot and does not prove transmutation and universal common ancestry. It proves that all living things were designed for specific and varying purposes. This is certainly true for humans. From everything we understand today, all the facts point to the strongest inference that intelligent design was the cause behind the origin of all life!
Then God said, “Let the land produce vegetation: seed-bearing plants and trees on the land that bear fruit with seed in it, according to their various kinds…Let the water teem with living creatures, and let birds fly above the earth across the expanse of the sky.” So God created the great creatures of the sea and every living and moving thing with which the water teems, according to their kinds, and every winged bird according to its kind…God made the wild animals according to their kinds, the livestock according to their kinds, and all the creatures that move along the ground according to their kinds… Then God said, “Let us make man in our image, in our likeness…So God created man in his own image, in the image of God he created him; male and female he created them. God blessed them and said to them, “Be fruitful and increase in number; fill the earth and subdue it…”
Genesis 1:20-28
Sources
1- Minkel, J.R., Human-Chimp Gene Gap Widens from Tally of Duplicate Genes. ScientificAmerican.com, http://www.sciam.com/article.cfm?articleID=9D0DAC2B-E7F2-99DF-3AA795436FEF8039. 19 December 2006.
2- Brittin, R. Divergence between Samples of Chimpanzee and Human DNA Sequences is 5%, Counting Indels, Proceedings of the National Academy of Sciences, USA 99 (2002): 13633–35.3-https://creation.com/decoding-the-dogma-of-dna-similarity
3- Uffelmann, E., Huang, Q.Q., Munung, N.S. et al. Genome-wide association studies. Nat Rev Methods Primers 1, 59 (2021). https://doi.org/10.1038/s43586-021-00056-9 https://www.nature.com/articles/s43586-021-00056-9
4- ICR “Human-Chimp Genetic Similarity: Is the Evolutionary Dogma Valid?” June 2011 https://www.icr.org/article/human-chimp-genetic-similarity-evolutionary
5- Birney, ENCODE Project Consortium,Bernstein, B. E., Birney, E., Dunham, I., Green, E. D., Gunter, C.,et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. DOI: 10.1038/ nature11247.
6- Gibney, E., Nolan, C. Epigenetics and gene expression. Heredity 105, 4–13 (2010).
7- Ruiz-Orera, J. et al. 2015. Origins of De Novo Genes in Human and Chimpanzee. PLoS Genetics. 11 (12): e1005721. Quoted from ICR article: “Genetic Gap Widens Between Humans and Chimps” Jeffery P. Tomkins, PH.D., Jan 2016.
8- Axe DD. Estimating the prevalence of protein sequences adopting functional enzyme folds. J Mol Biol. 2004 Aug 27;341(5):1295-315. DOI: 10.1016/j.jmb.2004.06.058. PMID: 15321723.
9- Oxford University Maths “Number of atoms in the universe” Nov 2015 https://educationblog.oup.com/secondary/maths/numbers-of-atoms-in-the-universe#:~:text=Our%20galaxy%2C%20the%20Milky%20Way,2.4%20%C3%97%201067%20atoms.
10- “Accuracy of rate estimation using relaxed clock models…” by Ho et. al., Article in Molecular Biology and Evolution, page 1355.
11- https://en.wikipedia.org/wiki/Species
12- Meyer, “Darwin’s Doubt” by Stephen C Meyer; 2013; pg. 106
13- Jamrozy, “Do Humans have the same DNA as bananas?” Jamrozy, Kelly; Medically Reviewed by
Dr. DHarshi Dhingra MD, with a specialization in Pathology. https://knowyourdna.com/humans-same-dna-as-bananas/#:~:text=Partially%2C%20yes.,Genome%20Research%20Institute%20in%202013.
14- Deziel, “Animals That Share Human DNA Sequences”, July 2018, Deziel, Chris https://sciencing.com/what-is-the-haploid-diploid-cell-number-for-a-monkey-12732203.html